Computational Modeling in f-element Chemistry
The 5f-orbitals and 6d-orbitals are well-known to engage in bonding motifs with multiconfigurational electronic structures and spin-orbit effects are frequently large. We perform state-of-the-art calculations that can be systematically improved on systems ranging from small molecules to large coordination complexes containing actinides. Our goal is to use higher-level methods than traditionally applied in the literature. We make synergistic comparisons between theory and highly accurate spectroscopic measurements. By computing properties using fully relativistic CASPT2 methods based on the Dirac equation (Dirac-CASPT2), we can understand the importance of the different relativistic effects as well as characterize the contributions from non-dynamic and dynamic correlation. Specifically, we study organoactinide complexes in collaboration with synthetic chemists, small molecules in collaboration with spectroscopists, and examine systematically chosen systems in collaboration with theoretical chemists developing new methods.
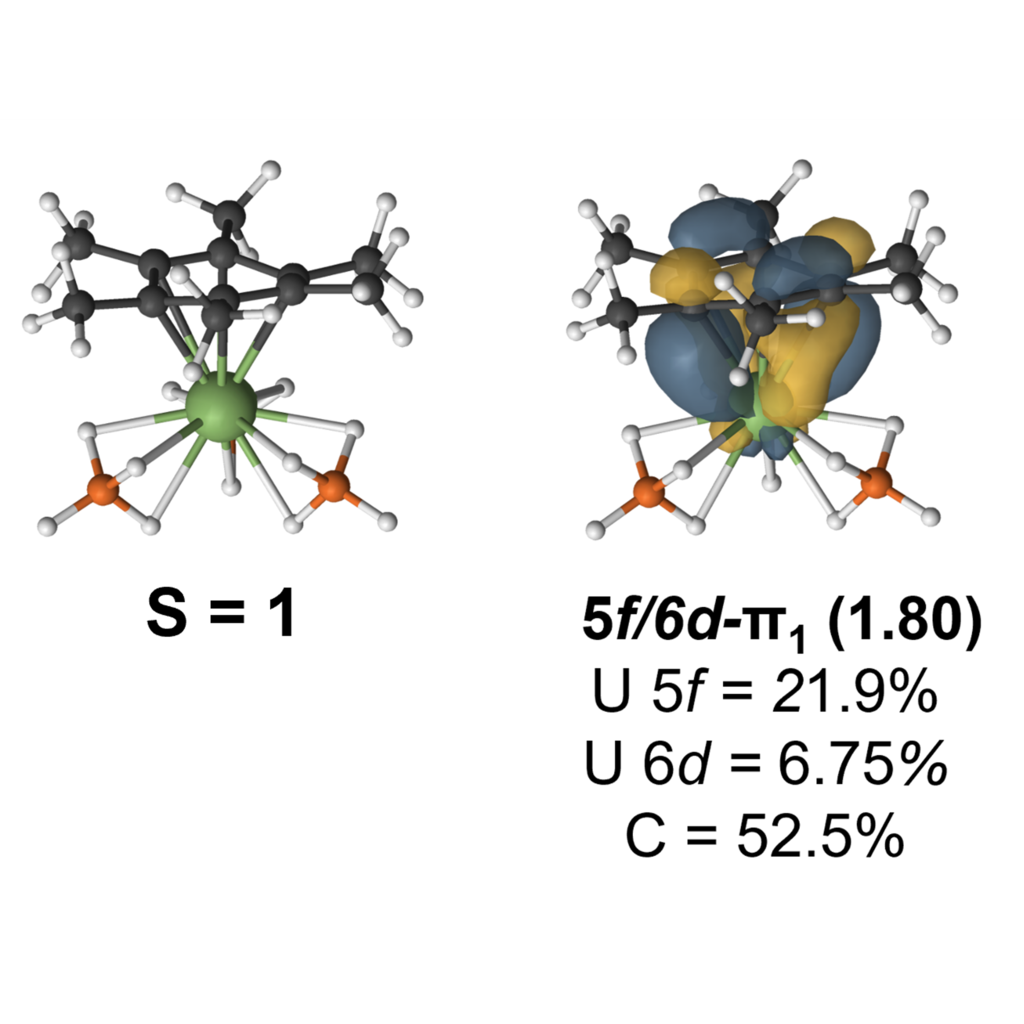
Actinide-Arene Interactions
The uranium-arene interaction energy shows that higher energy electron rich π-systems drive electrostatic contributions, while lower energy electron poor π-systems give rise to larger orbital contributions. Moreover, low occupancy U–arene π-bonding interactions dominate, while δ-bonding interactions are only found in the experimentally realized complex. Both DFT and multireference calculations on a reduced complex suggest oxidation state cannot be deduced from structural features alone.
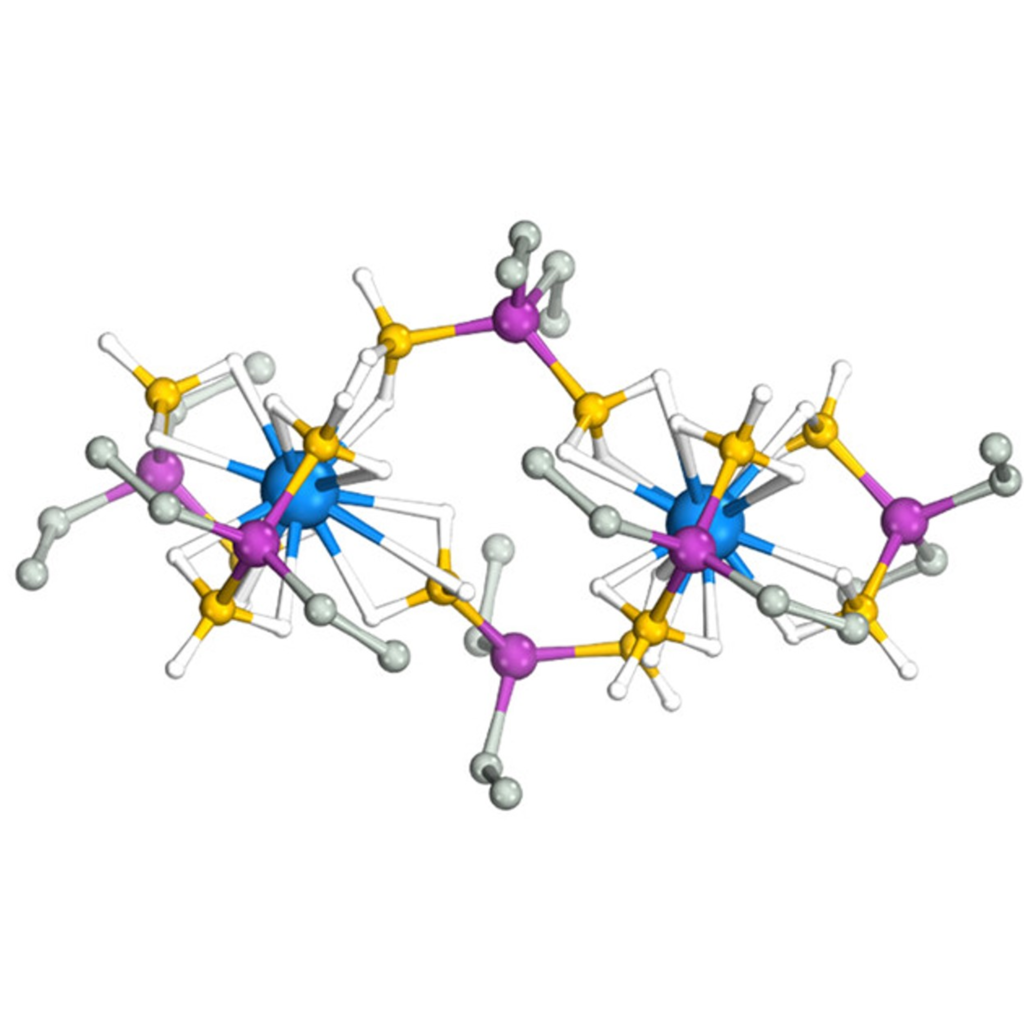
Quantifying Metal-Ligand Covalency
Experimental and computational studies quantify the measured effect of variations in metal-ligand covalency on the reactivity of phosphinodiboranate complexes with trivalent uranium and similarly sized lanthanides. Shorter-than-expected U−B distances indicate an increased covalency giving rise to measurable differences in solution deoligomerization reactivity when compared to isostructural complexes with similarly sized lanthanides.
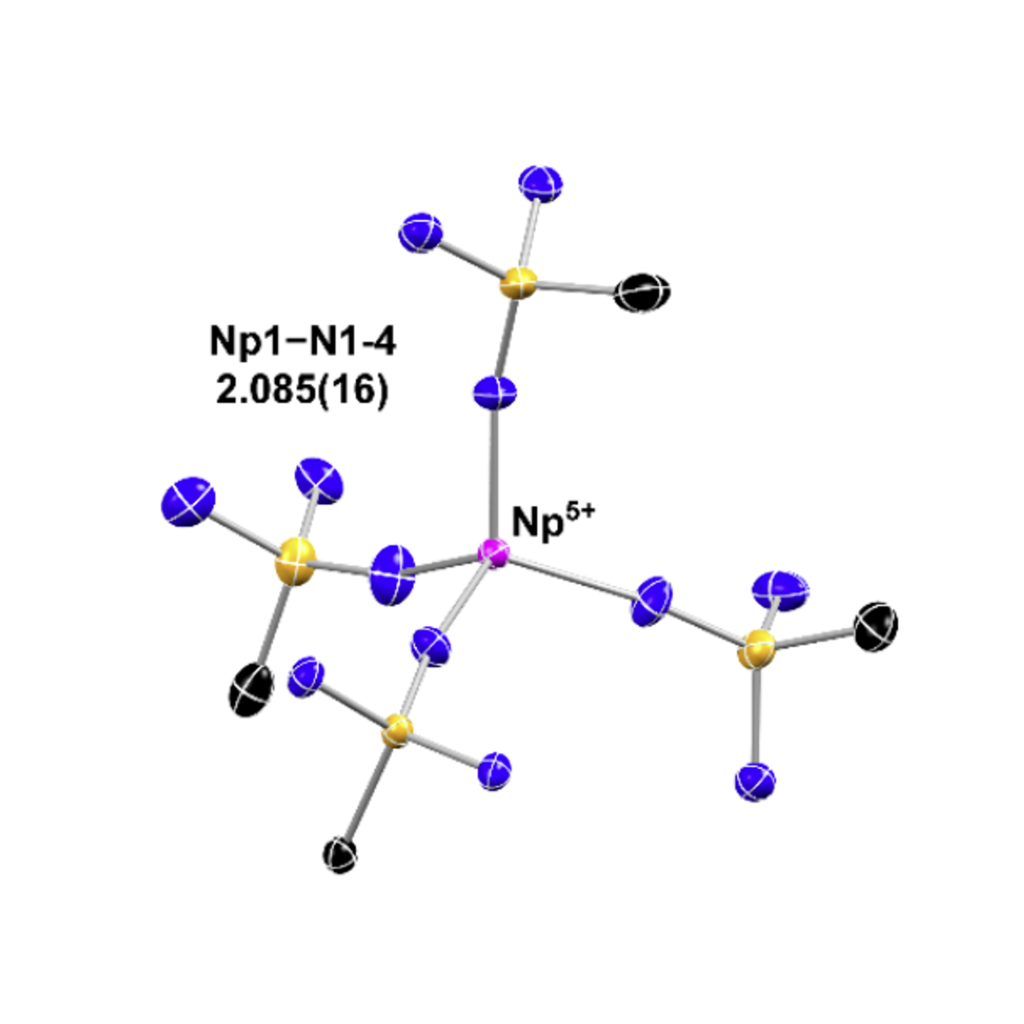
Zero-Field Splitting and Spin-Orbit Coupling
Using multireference methods, our group calculates the extent of spin-orbit coupling in actinide and lanthanide complexes. We are especially interested in novel oxidation states and transuranic systems. By employing a variety of methods that treat spin-orbit effects, ranging from state-interaction methods up to fully variational four-component methods, we can model the magnetic and spectroscopic properties accurately.
CASPT2 Molecular Geometries in Transition Metal Chemistry
Recent technological development of fully internally contracted gradients for second-order multireference complete active space methods (CASPT2) has enabled the geometry optimization of medium to large transition metal complexes. Our group has focused on using these methods to model the molecular geometries of iron spin-crossover complexes, copper corrole complexes, and Cr–Cr multiple bonds with CASPT2. While DFT geometries are often quite good for minima, this can break down when the electronic structure is best described with a multireference method. By performing geometry optimizations for the correct multireference ground state, one can ensure that the potential energy surface is described appropriately and avoid needing to take care with functional choice. This work is facilitated by implementations in the BAGEL program package, of which we are developers.
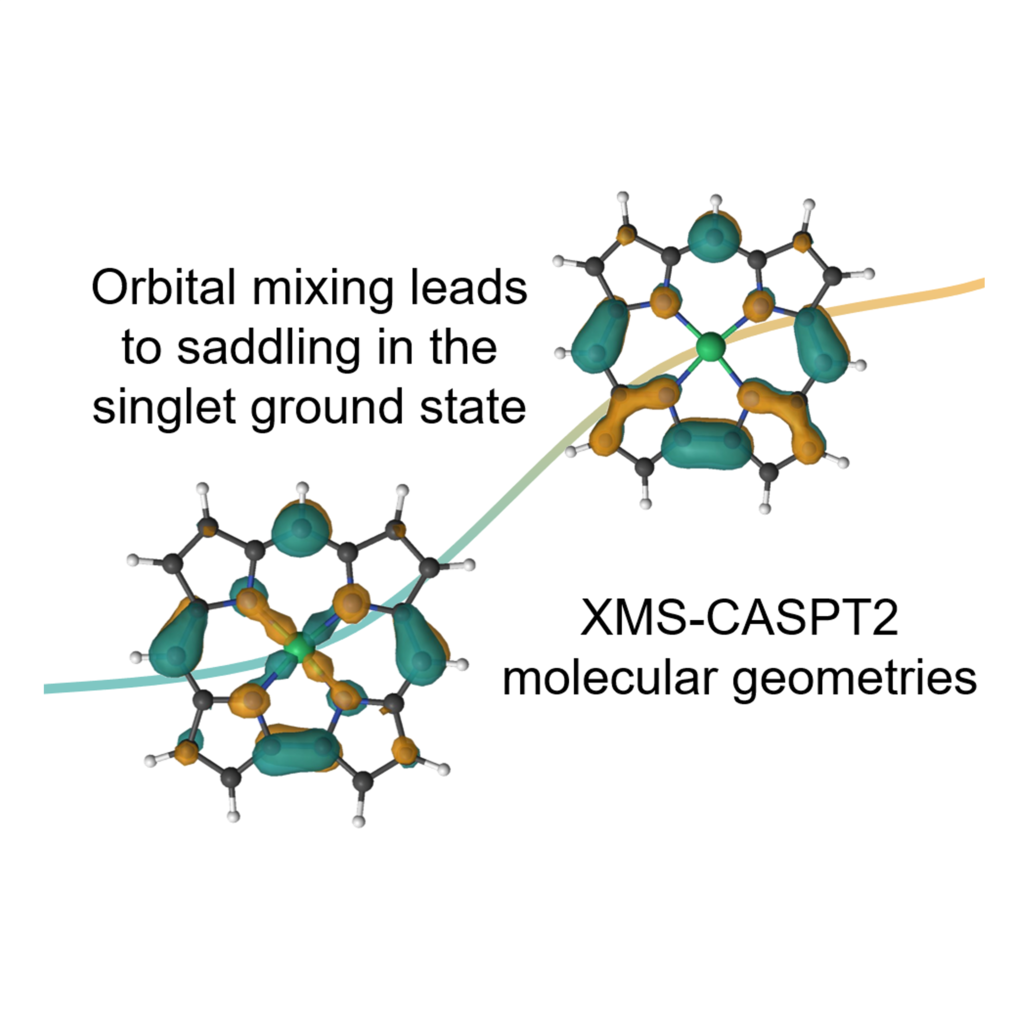
Molecular Geometry of Cu Corrole Complexes
Cu corroles are known for their unique multiconfigurational electronic structures in the ground state, which arise from the transfer of electrons from the π orbitals of the corrole to the d orbital of copper. While DFT provides reasonably good molecular geometries, the ground spin state is heavily influenced by functional choice. Using the XMS-CASPT2 method, the structure is optimized for the correct multiconfigurational state.
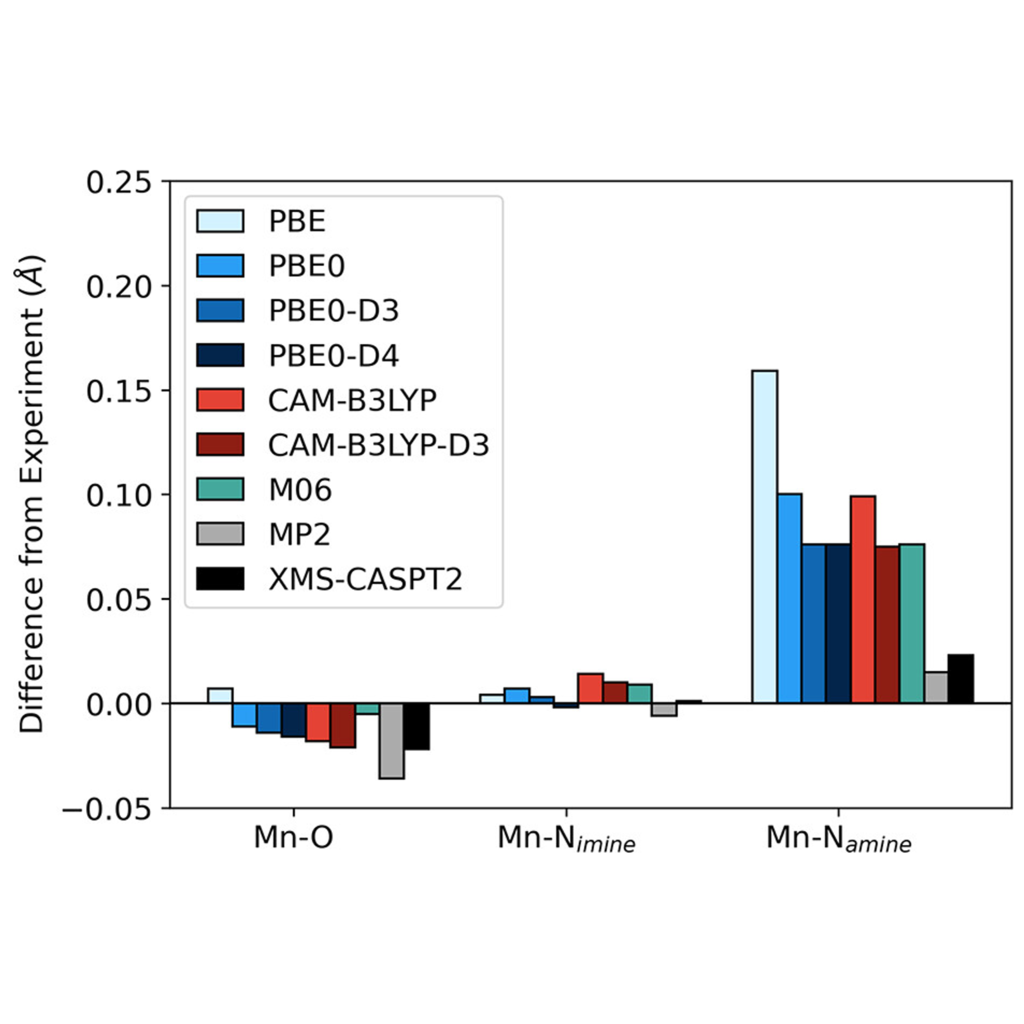
Importance of Dispersion in a Mn(III) Complex
For the geometry of the quintet high-spin state, density functionals significantly overestimate Mn–Namine bond distances, although the geometry for the triplet intermediate-spin state is well described. Comparisons with several wave function-based methods demonstrate that this error is due to the limited ability of commonly used density functionals to recover dispersion beyond a certain extent. XMS-CASPT2 offers a balanced approach, leading to better molecular geometries.
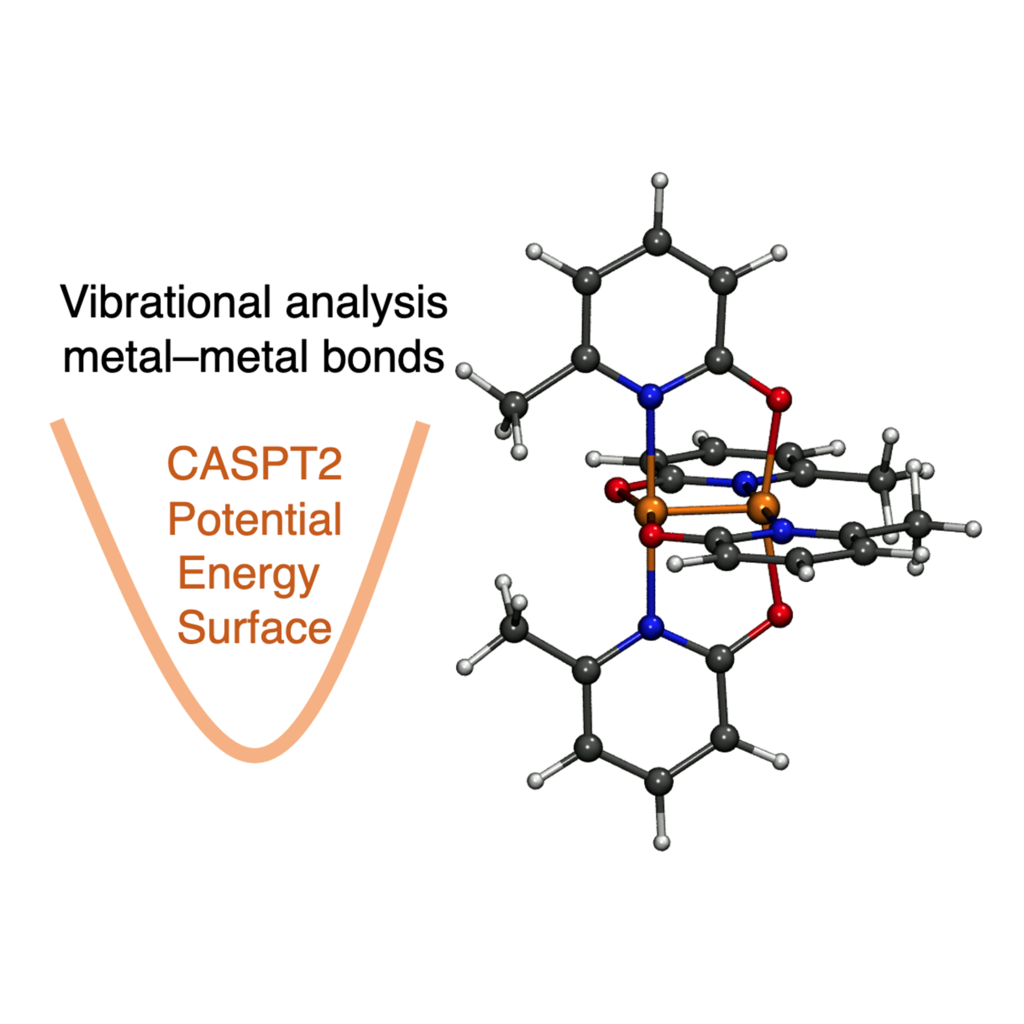
Vibrational Spectroscopy of a Cr-Cr Multiple Bond
Accurate computational vibrational analysis of the Cr–Cr bond in dichromium complexes using second-order multireference complete active space methods (CASPT2) allows for direct comparison with experimental spectroscopic data. This facilitates interpreting the low-energy region of the spectra and provides insights into the nature of the bonds themselves. Some Cr–Cr vibrational stretching modes suggest weaker bonding, even for so-called ultrashort Cr–Cr bonds, while others are in line with the bond distance.
Contributing Fundamental Understanding to Impact Global Challenges
Our group is motivated by a strong desire to contribute to large scall challenges in the environment. These problems cannot be solved alone and are often addressed in large collaborations. Within the NSF Center for Sustainable Polymers (NSF CSP), we perform computational modeling in combination with experiment to provide molecular-level insights to reaction mechanisms in order to guide the design of the next generation of catalysts. By means of a combination of density functional theory (DFT) and classical simulations, we address questions arising from experiments performed in the Center. We are currently collaboration with the Tolman group at Washington University in St. Louis, the Dauenhauer and Tonks groups at the University of Minnesota, and the Kalow group at Northwestern University. We are also members of the Environmentally Applied Refrigerant Technology Hub (EARTH), an NSF Engineering Research Center. EARTH is founded on four pillars: convergent research, diversity and culture of inclusion, engineering workforce development, and innovation ecosystem, which will impact the engineering and scientific communities, the HVACR industry, and society. Our role is to use quantum chemical methods for designing catalysts to make new refrigerants, to design novel sensors, and to improve separations.
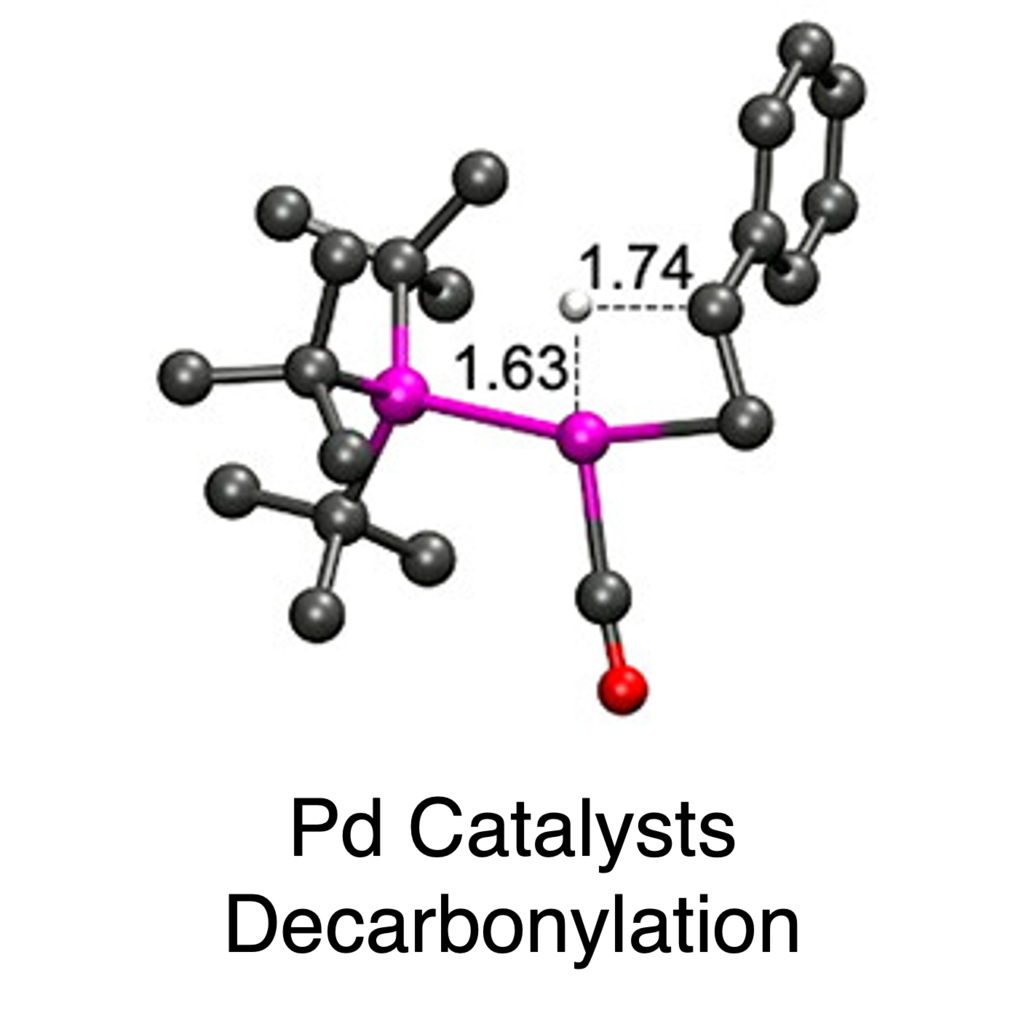
Ligand Effects on Decarbonylation
DFT calculations corroborated the experimental findings and provided insights into the ligand influences on reaction step barriers and transition state structures. Key findings include the following: a stable intermediate forms after chloride abstraction, from which β-hydride elimination is most affected by ligand choice, the low coordination number for the PtBu3 case lowers reaction barriers for all steps, and the trans disposition of two ligands for L = PPh3 contributes to the low efficiency for styrene production in that case.
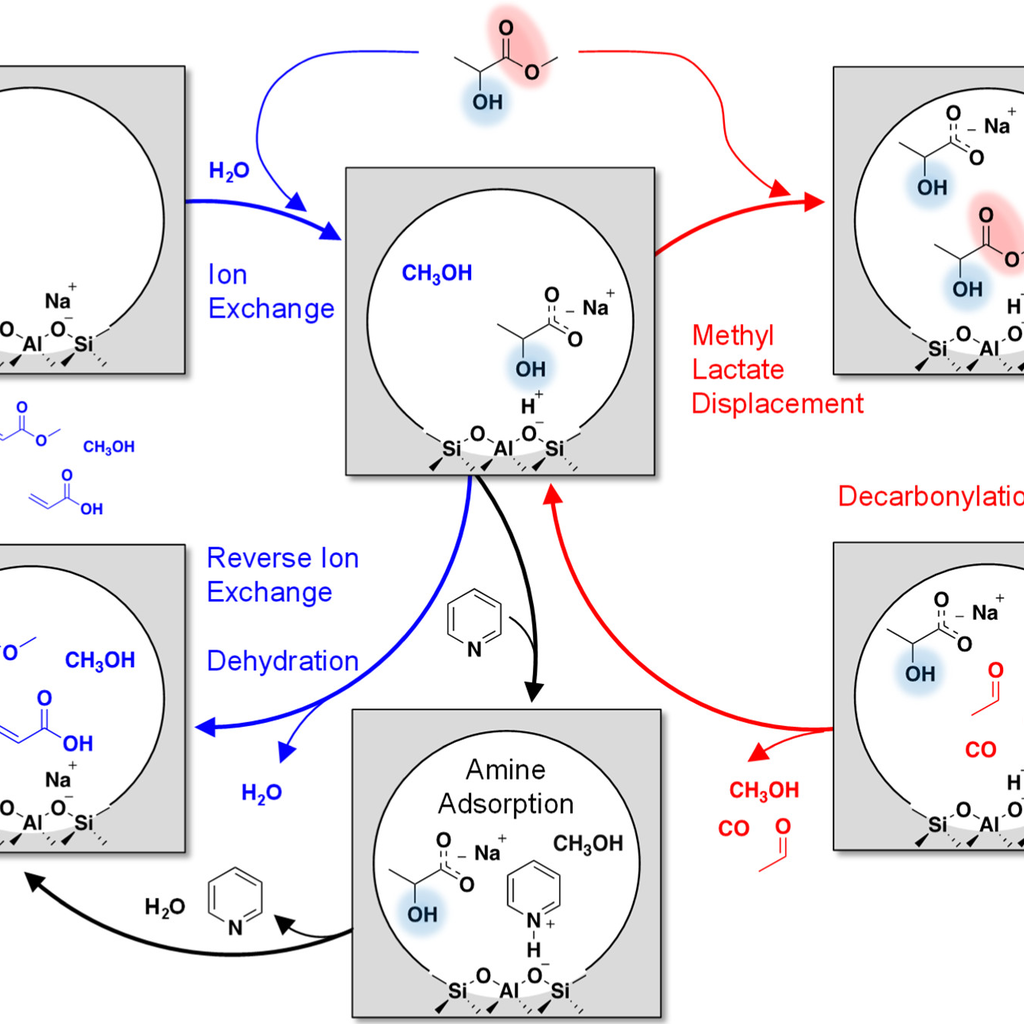
Selective Dehydration of Methyl Lactate to Acrylates
Co-feeding an inert and site-selective chemical titrant provides desirable selectivity tuning when titrant adsorption is favored over side reaction pathways on a solid acid catalyst. Here, a selectivity enhancement from 61 to 84 C % was demonstrated for methyl lactate dehydration to methyl acrylate and acrylic acid over a NaY zeolite catalyst using amines as the co-fed titrants to suppress side reactions on in situ-generated Brønsted acid sites (BASs). The effectiveness of BAS titration was evaluated by considering both the basicity and steric properties of the titrant molecule with the goal to maximize the selectivity enhancement.

Sulfurous Zeosils for Dehydra-Decyclization of THF
The computational results suggested that the nature of the Brønsted acid sites and the adsorption energetics (relative THF-acid site interaction energies) are distinct in each catalyst. Additionally, the protonation of THF can be improved with the addition of a water molecule acting as a proton shuttle, particularly in S-MFI. Overall, S-containing zeosils exhibited the ability to control reaction pathways and product distribution in dehydra-decyclization chemistry optimization within microporous zeolites, providing another alternative weak-acid catalytic material.